Sample sheet and demultiplexing
Quick links
Before pooling your libraries (and preparing your samplesheet), please make sure to check the index pooling guides.
How do I prepare my sample sheet?
At the end of the sequencing run, the raw data consist of images in the form of bcl files. The first data processing step is to generate fastq files. In most sequencing runs, several samples are combined with a set of multiplexing barcodes. It is called indexed sequencing and you can read more in the section below. Thus the demultiplexing step will generate a fastq file for each sample using the information from the sample sheet (which associate barcodes - singe or dual - to sample names).
- Download the template depending on your experiment: bulk template, 10X template or CRISPR template
- Fill out the template for this experiment, following the instructions below
- Place the excel file in the submission folder of your specific run
Filling out the bulk template
- name
- this column will define how the fastq files are named (an ending will be added with S1.fastq.gz, S2.fastq.gz, …)
- each name should be a one word (no space) without any special character (no #, +, -, % or other funny things - just letters, numbers, - and _ to separate). Please do not start with a number but rather with a letter (as this can be annoying afterwards in R)
- i7
- this is the sequence of the i7 barcode used for demultiplexing (we used to call it seqIndex1)
- it is typically 6 or 8 bp, depending on the multiplexing kit (e.g. NEB, etc.) - but it could be a different number depending on your kit
- if the index includes a UMI, please indicate it with the right number of Ns so that we know the structure and how we need to demultiplex
- e.g. GTTGACCTNNNNNNNNN (we sequence 17bp and extract the 9bp UMI when demultiplexing
- e.g. AGGTGTACNNNNNNNNNNNN (we sequence 20bp and extract the 12bp UMI when demultiplexing
- i5
- this is the sequence of the i5 barcode (we used to call it seqIndex2)
- if you use single-indexing, leave that column empty
- if the index includes a UMI, please indicate it with the right number of Ns so that we know the structure and how we need to demultiplex
- e.g. GTTGACCTNNNNNNNNN (we sequence 17bp and extract the 9bp UMI when demultiplexing
- e.g. AGGTGTACNNNNNNNNNNNN (we sequence 20bp and extract the 12bp UMI when demultiplexing
- contact
- when this is your project and you have prepared all samples and libraries, you just put your firstname
- when the pool is a merge of several libraries prepared by different people, or your prepared libraries from sets of samples coming from different people, you write the firstname of those people so we know which is which
- species - choose from the drop-down menu
- the species of the main genome for that sample - if not present, you can select other and let us know on email what species you would like us to add
- pipeline - choose from the drop-down menu
- if you just need fastq files, please select fastq
- if you want us to run one of our pipelines, please select the one you want. Info about the pipelines are here.
- if not present, please contact us
- dedup - choose from the drop-down menu
- by default this is without UMI
- if you have UMIs in your index and would like us to extract the UMIs at the demultiplexing stage as well as run a UMI-aware pipeline (e.g. CHOR), you can select with_UMI here
- control_name
- Only for pipelines that use IgG, KO or input controls (nf-cutandrun, nf-chip): indicate the name of the corresponding control (empty values are accepted)
Filling out the CRISPR template
- name
- this column will define how the fastq files are named (an ending will be added with S1.fastq.gz, S2.fastq.gz, …)
- each name should be a one word (no space) without any special character (no #, +, -, % or other funny things - just letters, numbers, - and _ to separate). Please do not start with a number but rather with a letter (as this can be annoying afterwards in R)
- i7
- this is the sequence of the i7 barcode used for demultiplexing
- it is typically 6 or 8 bp, depending on the multiplexing kit (e.g. NEB, etc.) - but it could be a different number depending on your kit
- i5
- this is the sequence of the i5 barcode (we used to call it seqIndex2 - but try to remove confusion here)
- if you use single-indexing, leave that column empty
- note: previously we have extracted this from the full sequence of the primer, checking that the sequence you indicated in i5 made sense with what we exactraced. But there might be a need to reverse-transcribed that one as compared to what you put before - we can talk about it if unclear.
- staggerSize
- if you did not use staggers, indicate 0
- if you used staggers, we suggest that you fill the last 4 columns with the full sequences of forward and reverse primers so that we can check the structure of the read (including the stagger sequence and size)
- contact
- when this is your project and you have prepared all samples and libraries, you just put your firstname
- when the pool is a merge of several libraries prepared by different people, or your prepared libraries from sets of samples coming from different people, you write the firstname of those people so we know which is which
- pipeline - choose from the drop-down menu
- if you want us to run one of our pipelines, please select the one you want. Info about the pipelines are here.
- if you select CRISPRESSO2, please make sure to fill out the CRISPRESS2-specific columns in your samplesheet.
- if not present, please contact us
- GUIDES - choose from the drop-down menu
- we list here the libraries that we have already formatted and worked with
- if not present, select NEW_LIBRARY and prepare a new library file following the instructions here
- CRISPRESSO2_XXX - required only when you select the CRISPRESSO2 pipeline. These columns follow the naming of the CRISPRESSO2 command parameters.
- BEAN_XXX - required only when you select MAGECK-BEAN pipeline. These columns follow the naming of the BEAN command parameters.
Filling out the 10X template
- If you have run 10X without hashes, just fill out the GEX table
- If you have run 10X with hasning, fill out both tables GEX and Hashing
- Just fill out values with the number of lines that you need
- Do not edit the headers
- Do not add additionnal information (you can email us instead if you want to provide some more info)
- Do not use any special character in the naming, nor space (in short, you can use letters and digits, possibly - and _)
What demultiplexing strategy do I need?
Standard demultiplexing
- We will use bcl2fastq with standard options
- bcl2fastq -R $RAWRUN -o . -r 16 -p 16 –sample-sheet samplesheet.csv –no-lane-splitting
Custom demultiplexing
- We will add a regular expression to specify the structure of the reads and indices and what we want to extract (e.g. parts of index and read), typically
- bcl2fastq -R $RAWRUN -o . -r 16 -p 16 –sample-sheet samplesheet.csv –no-lane-splitting –use-bases-mask REGULAR_EXPRESSION –create-fastq-for-index-reads –mask-short-adapter-reads=8
- In this case, you do need to specify in the request form the regular expression you want us to use with the –use-bases-mask option
- E.g. TTchem single-read single-indexed: Y*,I8Y11
- E.g. TTchem single-read dual-indexed: Y*,I8Y11,I8
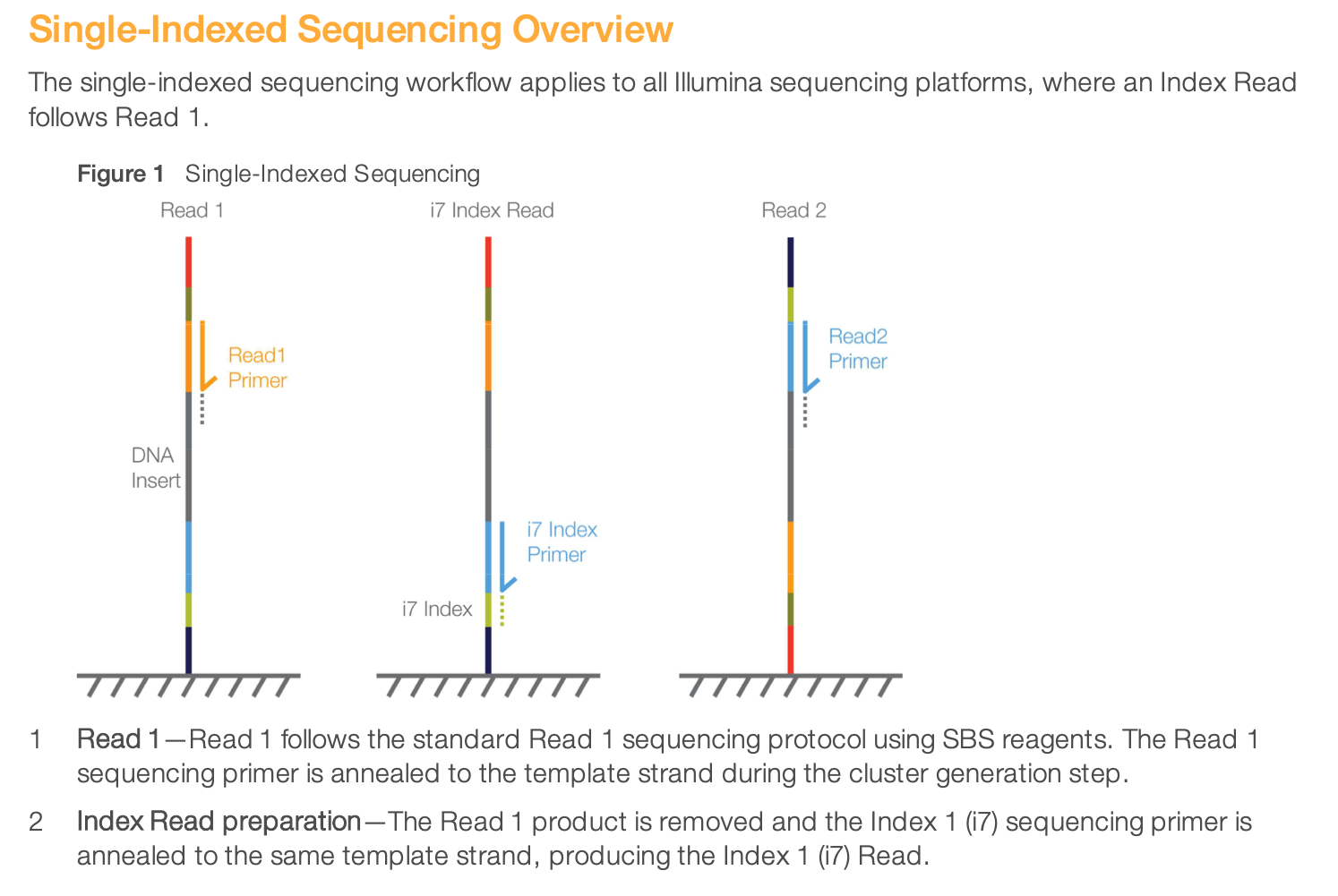
How does indexed sequencing work
To get all the details of indexed sequencing, you can read more from the Illumina documentation here. Let’s just show here the illustration of single and dual-indexed sequencing.
This is an illustration of single-indexed library sequencing.
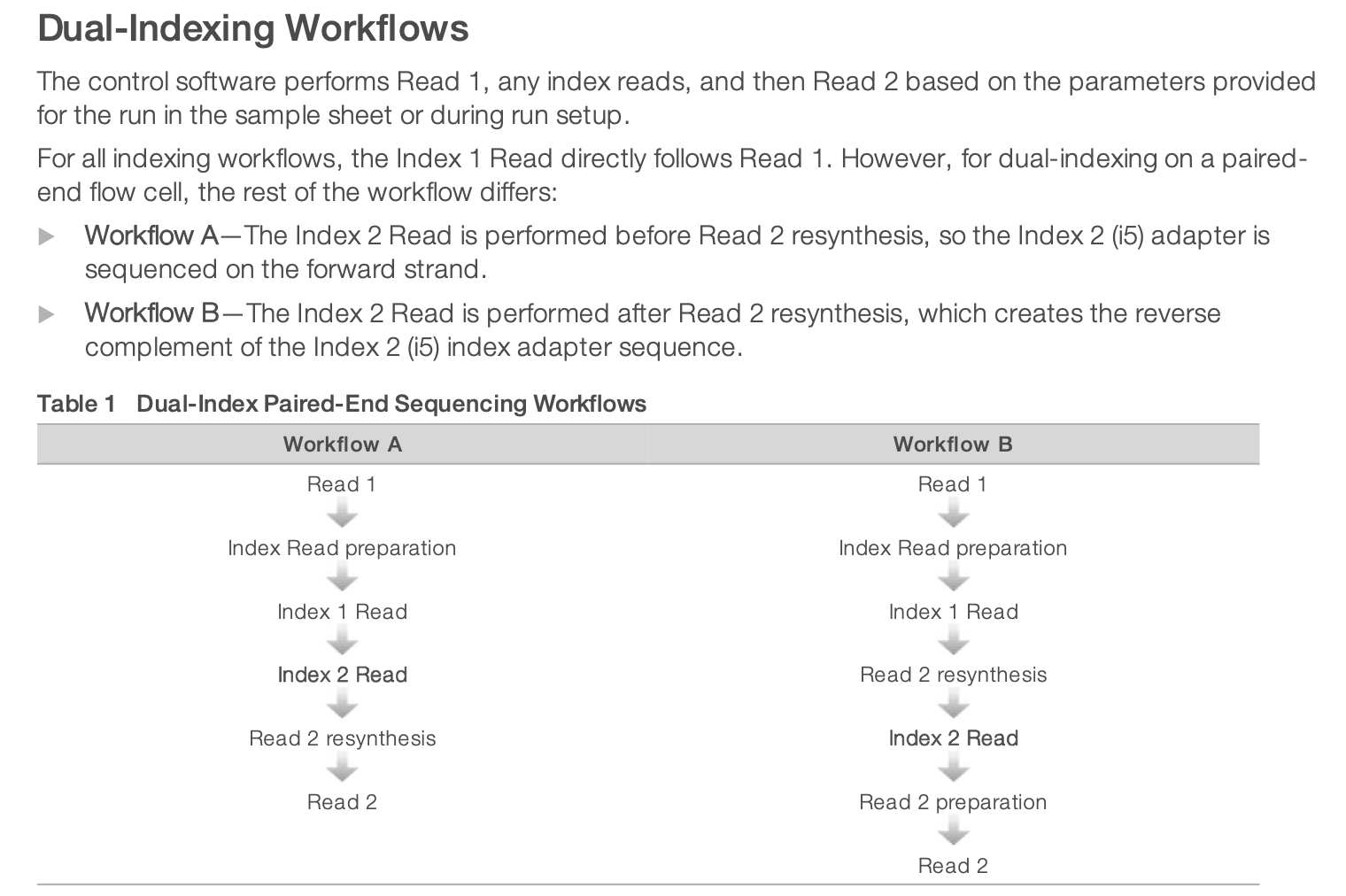
For dual-indexed library sequencing, please note that Illumona has 2 different workflows for different instruments. It means that in the indexing kit, you need to choose the column appropriate for NextSeq sequencing. It is using the Workflow B illustrated below.

Go back to the Genomics Platform home